
Transcriptome analysis of soybean roots in response to early symbiotic nitrogen fixation
-
摘要:
为挖掘大豆根中响应早期共生固氮过程中的关键基因,并探究这些关键基因的功能和代谢途径,本研究利用转录组学技术对不接种根瘤菌与接种根瘤菌1 d后的大豆根系材料进行RNA测序,通过对差异表达基因进行GO富集和KEGG富集分析来挖掘参与早期共生固氮过程中的关键基因,进一步筛选受根瘤菌调控的转录因子,再对其中富集数量最多的AP2/ERF家族成员进行进化分析,最后通过qRT-PCR验证其中部分基因的表达情况。研究共鉴定出824个响应根瘤菌侵染的差异表达基因(DEG),GO富集和KEGG富集分析结果表明,DEG主要富集在转录调控、激素相关以及次生代谢产物合成等通路上。进化分析结果,表明大豆中AP2/ERF家族成员可根据与MtERN1/2和LjERN亲缘关系的远近聚为三个分支,其中第Ⅰ分支可能在诱导根瘤菌侵染和根瘤器官发生中发挥作用。qRT-PCR结果表明,AP2/ERF转录因子在接种根瘤菌后确实受到不同程度地调控。本研究中发现的DEG为更好地理解早期共生固氮机制提供了候选分子。
-
关键词:
- 大豆 /
- 共生固氮 /
- 转录组 /
- AP2/ERF转录因子
Abstract:In order to explore the key genes in response to early symbiotic nitrogen fixation in soybean roots as well as the functions and metabolic pathways of these key genes, this study used transcriptomics technology to perform RNA sequencing on soybean root materials one day after with or without inoculation of rhizobia. The key genes involved in the early symbiotic nitrogen fixation process were explored by GO enrichment and KEGG enrichment analysis of differentially expressed genes, and the transcription factors regulated by rhizobia were further screened. The AP2/ERF family members with the largest number of enrichments were subjected to evolutionary analysis. Finally, the expression of some genes was verified by qRT-PCR. A total of 824 differentially expressed genes (DEGs) in response to rhizobium infection are identified. GO enrichment and KEGG enrichment analysis showed that DEGs are mainly enriched in transcriptional regulation, hormone-related and secondary metabolite synthesis pathways. Phylogenetic analysis showed that AP2/ERF family members in soybean could be clustered into three branches according to their genetic relationship with MtERN1/2 and LjERN. Among them, branch I may play a role in inducing rhizobium infection and nodule organogenesis. The results of qRT-PCR showed that the AP2/ERF transcription factor is indeed regulated to varying degrees after inoculation with rhizobia. The DEGs found in this study provide candidate molecules for a better understanding of the early symbiotic nitrogen fixation mechanism.
-
Keywords:
- soybean /
- symbiotic nitrogen fixation /
- transcriptome /
- AP2/ERF transcription factor
-
0 引 言
氮是植物生长发育所需的三大主要营养元素之一,其可利用性直接影响着作物生长状况和产量形成。大豆(Glycine max)作为全球广泛种植的作物之一,因其具有丰富的营养价值和极高的经济价值,因此,在我国的粮食和油料生产中占据着重要的地位。作为豆科植物的代表性作物,大豆可在土壤中氮含量不足的情况下,与根瘤菌共生形成根瘤并进行固氮,将大气中的氮气(N2)转化为可吸收的氨(NH3),为植株的生长发育提供氮源,进而提高大豆的产量[1]。此外,共生固氮也减少了农业生产过程中对化学氮肥的依赖,具有重要的生态和经济意义。
在共生固氮过程中,豆科植物根系会在低氮的土壤中分泌黄酮类化合物,吸引相容的根瘤菌附着于根毛上,黄酮类化合物被根瘤菌识别后会与结瘤蛋白(NodD)发生互作,引发根瘤菌结瘤基因(nodA,nodB,nodC)的表达,合成并释放结瘤因子(Nod factor ,NFs)——一种类脂几丁质寡糖(Lipo-chitooligosaccharides, LCOs)[2 − 5]。宿主植物根部通过结瘤因子受体(Nod factor receptors, NFRs)感知结瘤因子后激活了NF下游信号通路产生结瘤信号,引发表皮中的根瘤菌侵染过程:根毛膨大变形并将根瘤菌包裹住,随后植物细胞壁水解、侵染点周围的质膜内陷使根瘤菌进入植物细胞并形成管状的侵染线(Infection threads, ITs)[1,6];皮层细胞重新进入细胞周期并开始有丝分裂继而形成根瘤原基,与此同时根瘤菌随着侵染线伸长往皮层延伸最后定殖于根瘤原基中,并随根瘤原基一起发育为成熟根瘤进行固氮[7 − 10]。
在早期共生固氮过程中,一系列的信号传导和基因调控网络被激活,来调节根瘤菌侵染和根瘤的形成。根瘤菌释放的NF由NFR1(Nod factor receptor 1)和NFR5(Nod factor receptor 5)以及共受体SYMRK(Symbiosis receptor-like kinase)形成的受体复合物所感知,导致根毛表皮细胞膜电位去极化引发膜内钙离子流和核内钙离子震荡,产生钙峰-结瘤信号激活依赖钙和钙调蛋白的蛋白激酶(Calcium- and calmodulin-dependent protein kinases, CCaMK)[6,11 − 13]。活化的CCaMK通过与转录因子CYCLOPS互作将其磷酸化,磷酸化的CYCLOPS以序列特异性的方式与DELLAs和NSP(Nodulation signaling pathway)1/2相互作用,反式激活NIN(Nodule inception)的表达[14 − 17]。此外,CYCLOPS还能与ERN1(Ethylene responsive factor required for nodulation1)启动子特异结合,并与CCaMK形成复合物正向调节ERN1转录[18]。同时,NSP1能与NSP2互作形成异源二聚体并通过顺式元件AATTT直接与ENOD11(Early nodulin 11)启动子结合,ENOD11是一个早期结瘤蛋白,在根瘤菌侵染前和侵染阶段的根和根瘤组织中转录[19 − 20]。ERN1、ERN2也能与ENOD11启动子特异性结合激活其表达,而ERN3抑制这种依赖ERN1、ERN2的ENOD11转录激活[21]。NIN是一个核心转录调控因子,有研究表明:NIN参与根瘤菌侵染期间的侵染线形成、根瘤菌定殖等过程以及根瘤器官发生过程[22 − 24]。尽管已经有大量研究关注了大豆-根瘤菌共生过程中的分子机制,但参与调控共生固氮的基因中仍存在大多数关键基因未被鉴定。
当前,转录组学技术具有高通量性和高灵敏度的特点,已成为挖掘大豆响应共生固氮关键基因的有力工具。利用转录组学技术探索在接种根瘤菌后的特定时间节点大豆基因的mRNA丰度,有助于更全面深入地理解大豆与根瘤菌之间的互作机制。Hayashi等[25]通过用野生型根瘤菌或无效突变体(nodC-)菌株接种根,对结瘤区进行转录组分析鉴定出
2915 个被根瘤菌产生的Nod因子信号特异性调控的基因。Yuan等[26]采用RNA-seq方法研究了大豆根系在5个不同接种后时间点(0.5、7-24 h,5、16和21 d)以及与两种不同菌株(113-2和USDA205)共生的差异表达基因,发现这些基因主要编码大豆抗性蛋白、NF相关蛋白、结瘤蛋白和免疫防御蛋白,以及与类黄酮/黄酮/黄酮醇生物合成和植物-病原体相互作用的有关蛋白质。祁平等[27]对根瘤菌接种12 h后大豆根系进行转录组测序,发现16个编码损伤诱导短肽(Wound-Induced Peptide, WIP)的基因在根瘤菌侵染后被诱导表达。本文旨在利用转录组学技术,通过分别比较大豆根在不接种根瘤菌与接种根瘤菌1 d后的基因表达谱,分析根瘤菌侵染后植物根部的表达变化,探究参与共生过程的关键基因和代谢途径,进而为揭示早期共生固氮的分子机制提供理论依据。通过这项研究,有助于研究和了解大豆根中的表达模式和调控,为提高大豆产量和氮素利用效率提供新的理论和实践基础。
1 材料与方法
1.1 实验材料
本研究所用大豆(Glycine max)材料为栽培品种Williams 82和慢生型根瘤菌(Bradyrhizobium japonicum)USDA110,均来自中国科学院大豆分子设计育种重点实验室。
1.2 材料预处理与收集
将籽粒饱满、大小基本一致、表面无裂缝的大豆种子经氯气(100 ml NaClO + 4 ml浓HCl)灭菌16 h后于通风橱中吹走剩余氯气,移至铺有湿润无菌滤纸的培养皿中于23 ℃黑暗条件下放置3 d使其萌发出芽。待种子萌发后转移到装有无菌的蛭石和珍珠岩(体积比为2∶1)的盆中,于温度为28 ℃、相对湿度为50%、光照条件为14 h光照/10 h黑暗交替进行的气候室中恒温培养。USDA110(抗性为氯霉素)在28 ℃摇床活化至菌液OD600值为0.9~1.2,将菌液
6000 r·min−1离心5分钟后倒掉上清液,再用无氮营养液稀释至OD600值为0.1左右;处理组在植物根部附近使用5 ml移液枪接种10 ml根瘤菌菌液,对照组则用10 ml无氮营养液处理;根瘤菌接种1 d后对植物根系进行取样,该时间点是大豆根毛被根瘤菌侵染的关键时期,此时有侵染线形成且皮层细胞开始分裂[26]。将样品用液氮速冻后放在−80 ℃冰箱保存。每个处理都有3个生物学重复。1.3 RNA提取和检测
使用Trizol试剂盒(Invitrogen , CA , USA)按照说明书提取总RNA,再用Bioanalyzer 2100和RNA 1000 Nano LabChip Kit(Agilent, CA, USA)进行总RNA质量和纯度分析。总RNA经质检合格后方可用于后续文库构建和测序。
1.4 文库构建和测序
从提取的总RNA中富集mRNA并随机打断成短片段,随后利用随机引物(Random hexamers)根据这些片段合成cDNA的第一条链,再添加缓冲液、dNTPs、RNaseH和DNA聚合酶I合成cDNA的第二条链。接着对合成的双链cDNA进行纯化并将其粘性末端修复为平末端,同时在3´端加碱基A和测序接头,经过片段选择后进行PCR扩增,最终构建出用于测序的cDNA文库。在确认文库质量达标后,使用Illumina Novaseq™ 6000仪器进行测序。文库的构建和转录组测序均由联川生物科技有限公司负责执行。
1.5 测序数据处理
1.5.1 测序数据预处理
通过Cutadapt软件过滤掉(文库构建过程中引入的)测序接头和(由于测序仪自身误差产生的)不合格的测序数据,从而对原始数据进行预处理得到有效数据;不合格的序列主要包括:含有超过5%比例N(N表示无法确定碱基信息)的reads、低质量reads(质量值Q≤10的碱基数占整个read的比例超过20%)。再通过使用FastQC软件统计原始测序量、有效测序量、Q20、Q30、GC含量对整体测序质量进行分析。
1.5.2 与参考基因组进行比对
使用HISAT2.0软件将测序有效数据与大豆参考基因组(https://genome.jgi.doe.gov/pages/dynamicOrganismDownload.jsf?organism=Gmax)进行比对,根据比对的结果使用StringTie软件来拼接与合并转录本并以FPKM(Fragments Per Kilobase of exon model per Million mapped reads)值量化基因表达水平。同时根据GO数据库(http://geneontology.org)和KEGG数据库(http://www.kegg.jp/kegg)等数据库中的基因组注释文件对基因进行注释。
1.6 差异表达基因的富集分析
使用edgeR软件对两个处理下共6个样品中表达的基因进行差异表达鉴定并对其进行GO和KEGG功能富集分析,差异表达基因(Differentially expressed gene, DEG)的筛选条件为P<0.05且|log2foldchange|≥1,FC(fold change)为差异倍数,然后通过使用R语言制作火山图、散点图等图形对差异表达结果进行可视化。以log2(fold change)为横坐标,-log10(P value)为纵坐标绘制火山图来了解DEG的整体分布情况。采用ggplot2分别绘制DEG GO富集和KEGG富集的分析结果的散点图。
1.7 转录因子分析以及AP2/ERF转录因子家族在不同组织中的表达模式分析
鉴于转录因子在早期共生固氮中的重要作用,筛选出所有受根瘤菌侵染调控差异显著的转录因子并与植物转录因子数据库PlantTFDB(https://planttfdb.gao-lab.org/index.php)进行比对统计转录因子的数量和类型。将差异表达的36个AP2/ERF家族成员进一步与植物转录因子数据库PlantTFDB中已鉴定的大豆AP2/ERF各亚家族(AP2、ERF、DREB、RAV和Soloist)中的成员进行比对并分类。为了进一步了解响应早期共生固氮的AP2/ERF转录因子各亚家族成员在根、根瘤、地上部、叶等不同组织中的表达模式,利用已公开的植物RNA-seq在线数据库(https://plantrnadb.com/)[28]下载这些家族成员基因在不同组织中的相关表达数据并使用GrapPad Pism 8制作基因表达热图。
1.8 AP2/ERF转录因子家族进化分析
从Phytozome数据库(https://phytozome-next.jgi.doe.gov/)中获取筛选出的36个大豆AP2/ERF转录因子家族成员的氨基酸序列,同时从NCBI数据库中下载蒺藜苜蓿ERN1/ERN2和百脉根ERN的氨基酸序列。随后,使用MEGAX软件对这些序列进行多序列比对,并采用邻接法(Neighbor-Joining)构建系统发育树,设定p-distance作为遗传距离计算方法,并执行了
1000 次bootstrap自展值计算以提高发育树的可靠性,同时选择了成对删除空位选项,对于其他参数则保留了软件的默认设置。1.9 qRT-PCR分析
使用不接种根瘤菌和接种根瘤菌1 d后的根组织样品提取RNA,再以ELF-1b基因作为内参对10个差异表达的AP2/ERF转录因子家族成员基因进行qRT-PCR验证。采用Trizol(Invitrogen,CA,USA)法提取植物总RNA,用Trans‐Script® All-in-One-Step First-Strand cDNA Synthesis Super Mix for qPCR (One-Step gDNA Removal)(北京全式金生物技术有限公司)试剂盒反转录成cDNA,在NCBI网站(https://www.ncbi.nlm.nih.gov/tools/primer-blast/)上设计引物(表1)后由生工生物工程(上海)股份有限公司合成引物序列,使用以下反应体系(表2)和程序(表3)进行qRT-PCR反应,用Excel整理内参及基因表达数据,基因转录水平用相对定量法进行计算后使用软件GrapPad Pism 8进行统计分析并作图。
表 1 qRT-PCR引物名称及序列Table 1 Primer name and sequence of qRT-PCR引物名称
Primer name序列(5'-3')
Sequence(5'-3')引物名称
Primer name序列(5'-3')
Sequence(5'-3')qRT-Gm01G225000-F GGAGGGAATGCGAGACAGAA qRT-Gm03G111700-F TTGAGATACCCGTAACGACGC qRT-Gm01G225000-R TGTTGTTGTGGGGCAATTCG qRT-Gm03G111700-R CTCCGCCCAATTCTCTGTCA qRT-Gm16G040000-F CCAAACCTGCAGCAACAGAAAT qRT-Gm02G080200-F TAGCATCTCAAAGGGTGTCCTG qRT-Gm16G040000-R ATCTCAGCAACCCATCTCCC qRT-Gm02G080200-R TCCGTGAACCGCATAAACTCA qRT-Gm17G210500-F GGGAGTAAGACAGCGCCAAT qRT-Gm03G162500-F TCATGGGAAGAGCTGTTTTCG qRT-Gm17G210500-R GCAGCCTCTGCTGTCTCAAA qRT-Gm03G162500-R ATTTTTCTTCAACGTGGCCGT qRT-Gm07G091100-F AGGCAAACTCAGAAACAAGGAA qRT-Gm12G073300-F GACAGCGAAGTAGAAGCTGCAAG qRT-Gm07G091100-R GTGGCATCAGGGTCAGTGTA qRT-Gm12G073300-R CTGCCCATTGGGAAAGAAATTAGG qRT-Gm17G169800-F AGGGTGAAACTGAGACGACG qRT-ELF1b-F GTTGAAAAGCCAGGGGACA qRT-Gm17G169800-R GAAAACGGCGGTGTCGTAAG qRT-ELF1b-R TCTTACCCCTTGAGCGTGG qRT-Gm05G186700-F AACGACGTTGACCAGAGTTACA qRT-Gm05G186700-R AAAGCAGAGATCATGGCTGACA 表 2 qRT-PCR反应体系Table 2 qRT-PCR reaction system试剂
Reagent用量
Consumption/μLcDNA 1 Primer F 0.2 Primer R 0.2 ChamQ Blue Universal SYBR qPCR Master Mix 5 ddH2O 3.6 表 3 qRT-PCR反应程序Table 3 qRT-PCR reaction procedure温度
Temperature/ ℃时间
Time/s95 30 95 10 $\Biggr\} $ 40 cycles 60 30 2 结果与分析
2.1 测序数据质量分析
为了挖掘大豆根中响应早期共生固氮的关键基因,收集根瘤菌USDA110侵染1 d的根系材料,同时以水处理为对照,每组3个生物学重复,共6个样品进行转录组测序,获得
86149352 条原始数据;经预处理过滤掉不合格的序列后得到83943518 条有效数据,有效数据占比为96.7% ~ 97.5%,质量值≥30的碱基占比为97.0% ~ 97.3%,GC含量占比均为44.5%(表4)。数据表明测序质量较高,可用于后续分析。表 4 测序数据质量分析Table 4 Quality analysis of sequencing data样品编号
Sample code原始数据
Raw reads有效数据
Valid reads有效数据占比
Valid ratio/%质量值≥30的碱基占比
Q≥30/%GC含量占比
GC content/%NN_1d_CK1 41981696 40909042 97.44 96.98 44.5 NN_1d_CK2 42443112 41316398 97.35 97.28 44.5 NN_1d_CK3 43652536 42547784 97.47 97.32 44.5 NN_1d_Bj1 41971824 40604942 96.74 97.31 44.5 NN_1d_Bj2 41274128 40152166 97.28 97.25 44.5 NN_1d_Bj3 44167656 43034476 97.43 97.33 44.5 2.2 差异表达基因的富集分析
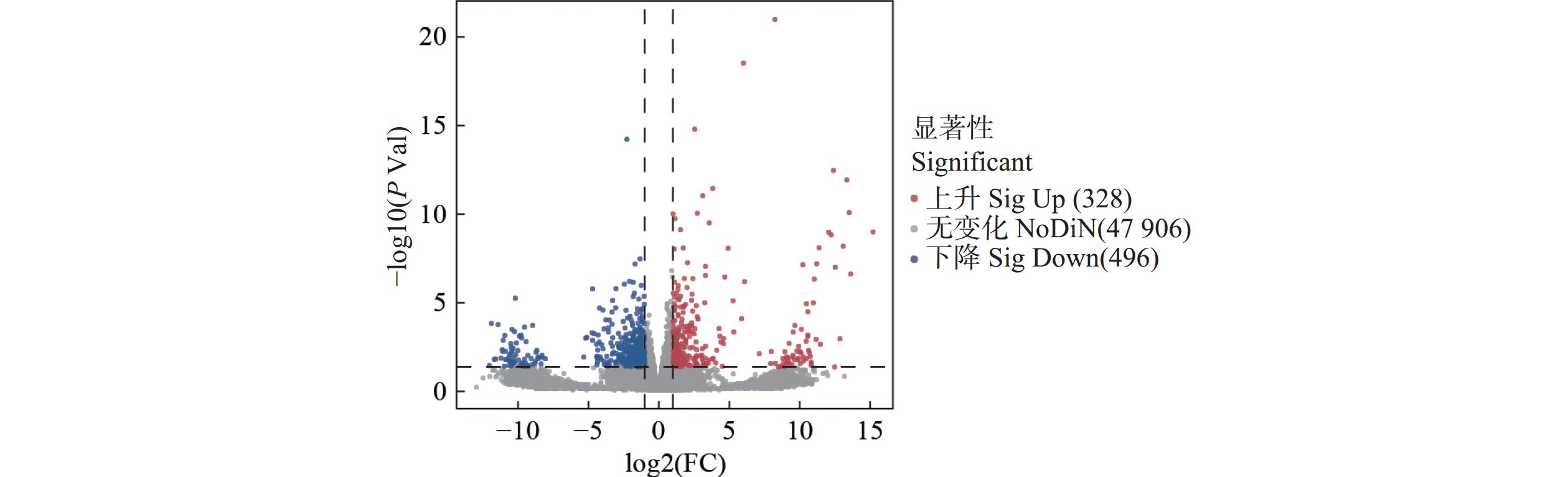
以FC≥2或FC≤0.5且P<0.05为标准,共筛选出了824个DEGs,其中有328个基因上调表达,496个基因下调表达(图1)。328个上调表达基因中有43个基因的转录本在不接种的根细胞中未发现,表明这些基因对根瘤菌接种有特异性响应(附表1)。在这些差异表达基因中,Glyma.01G028500、Glyma.09G187000、Glyma.02G311000、Glyma.01G241900、Glyma.06G184400这5个基因已经报道过在共生固氮中的作用。Glyma.09G187000(FW2.2-like 1,GmFWL1)能响应根瘤菌侵染在根毛中表达并随后在根瘤发育过程中被强烈诱导表达,可能参与植物细胞对根瘤菌侵染响应所需的细胞重塑程序,如参与皮质细胞分裂的起始和侵染线渗透到根瘤原基的过程,RNAi沉默GmFWL1可显著减少根瘤数量并减小细胞核大小[29]。Glyma.01G241900(Nucleolar/Mitochondrial protein involved in nodulation a,GmNMNa)在根瘤菌侵染期间特异性表达,其编码定位于核仁和线粒体、功能未知的蛋白质,RNA介导的GmNMNa基因沉默导致结瘤数量减少、侵染细胞中的类菌体数量减少以及类菌体中聚-β-羟基丁酸酯(Poly-β-hydroxybutyrate,PHB)的积累明显减少[30]。
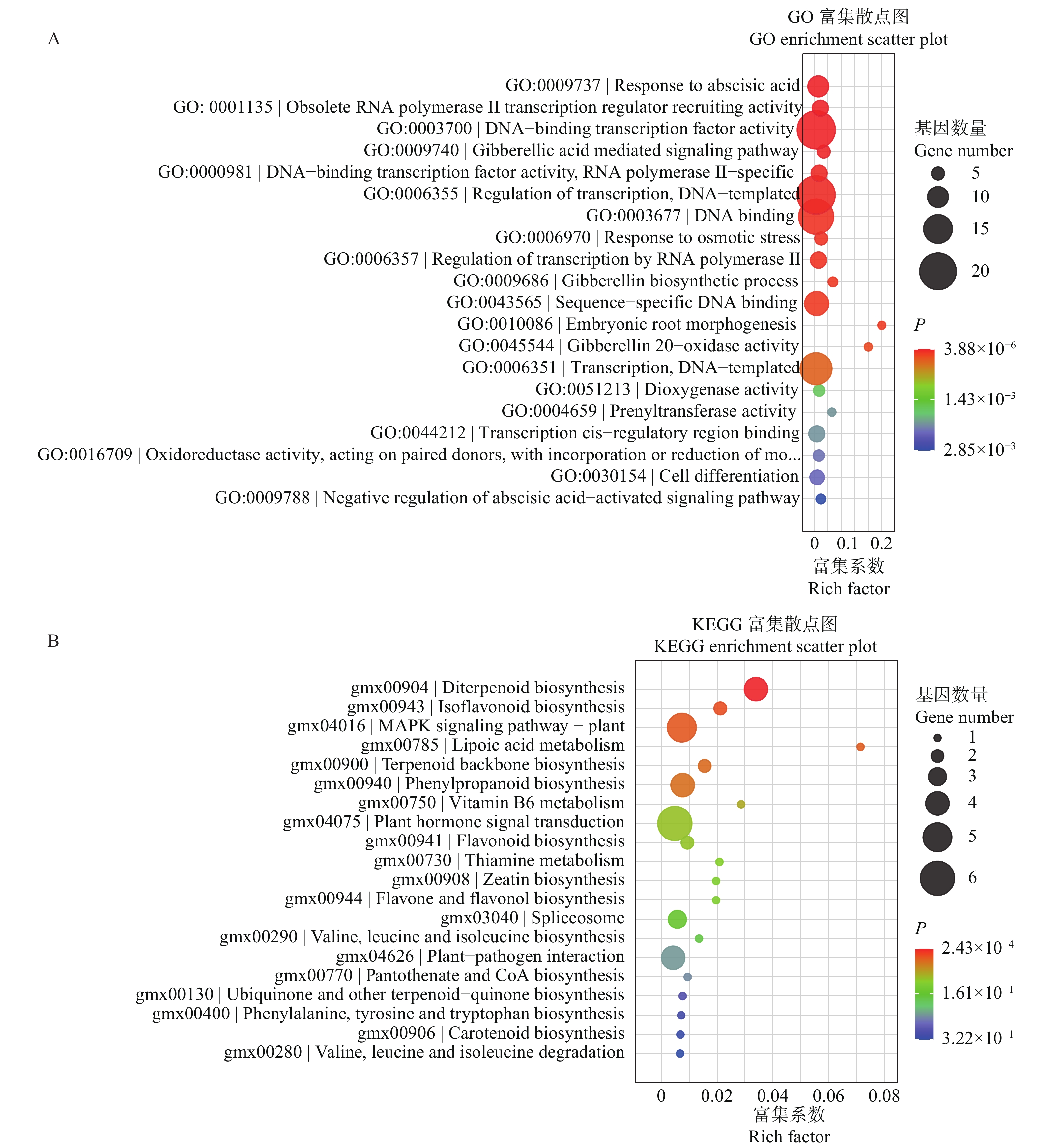
为了解在大豆—根瘤菌早期共生互作过程中基因表达的特点,对差异表达基因(DEGs)进行GO富集分析(图2A),结果表明DEGs在与转录调控和激素相关的GO条目中显著富集,如DNA结合转录因子活性(DNA−binding transcription factor activity)、以DNA为模板的转录调控(regulation of transcription,DNA−templated)、脱落酸响应(response to abscisic acid)、赤霉酸介导的信号通路(gibberellic acid mediated signaling pathway)。此外,DEGs还富集在胚根形态发生(embryonic root morphogenesis)、细胞分化(cell differentiation)等与皮层细胞分裂有关的GO条目和赤霉素生物合成过程(gibberellin biosynthetic process)、次级代谢产物生物合成过程(secondary metabolite biosynthetic process)等代谢产物合成的GO条目中。
![]() 注:Rich factor表示位于GO条目中的差异基因个数与总基因数的比值,其大小与富集程度呈正比,颜色与P值大小有关。Note: Rich factor represents the ratio of the number of differentially expressed genes in the GO entries to the total number of genes, which is proportional to the degree of enrichment, and the color is related to the size of the P value.图 2 差异表达基因的GO富集散点图(A)和差异表达基因的KEGG富集散点图(B)Fig. 2 GO enrichment scatter plot of differentially expressed genes (A) and KEGG enrichment scatter plot of differentially expressed genes (B)
注:Rich factor表示位于GO条目中的差异基因个数与总基因数的比值,其大小与富集程度呈正比,颜色与P值大小有关。Note: Rich factor represents the ratio of the number of differentially expressed genes in the GO entries to the total number of genes, which is proportional to the degree of enrichment, and the color is related to the size of the P value.图 2 差异表达基因的GO富集散点图(A)和差异表达基因的KEGG富集散点图(B)Fig. 2 GO enrichment scatter plot of differentially expressed genes (A) and KEGG enrichment scatter plot of differentially expressed genes (B)通过KEGG富集分析进一步分析差异表达基因所影响的代谢通路(图2B),结果表明DEGs主要富集在植物激素信号传导(Plant hormone signal transduction)、MAPK信号通路(MAPK signaling pathway−plant)、植物-病原体互作(Plant−pathogen interaction)以及各种代谢产物合成和代谢通路上,代谢产物合成通路主要包括二萜类化合物生物合成(Diterpenoid biosynthesis)、异黄酮生物合成(Isoflavonoid biosynthesis)、萜类化合物骨架生物合成(Terpenoid backbone biosynthesis)、苯丙烷类化合物生物合成(Phenylpropanoid biosynthesis)、类黄酮生物合成(Flavonoid biosynthesis),代谢产物代谢通路包括硫辛酸代谢(Lipoic acid metabolism)、维生素B6代谢(Vitamin B6 metabolism)和硫胺素代谢(Thiamine metabolism)。
2.3 早期共生固氮中转录因子的分析和AP2/ERF家族组织表达模式分析
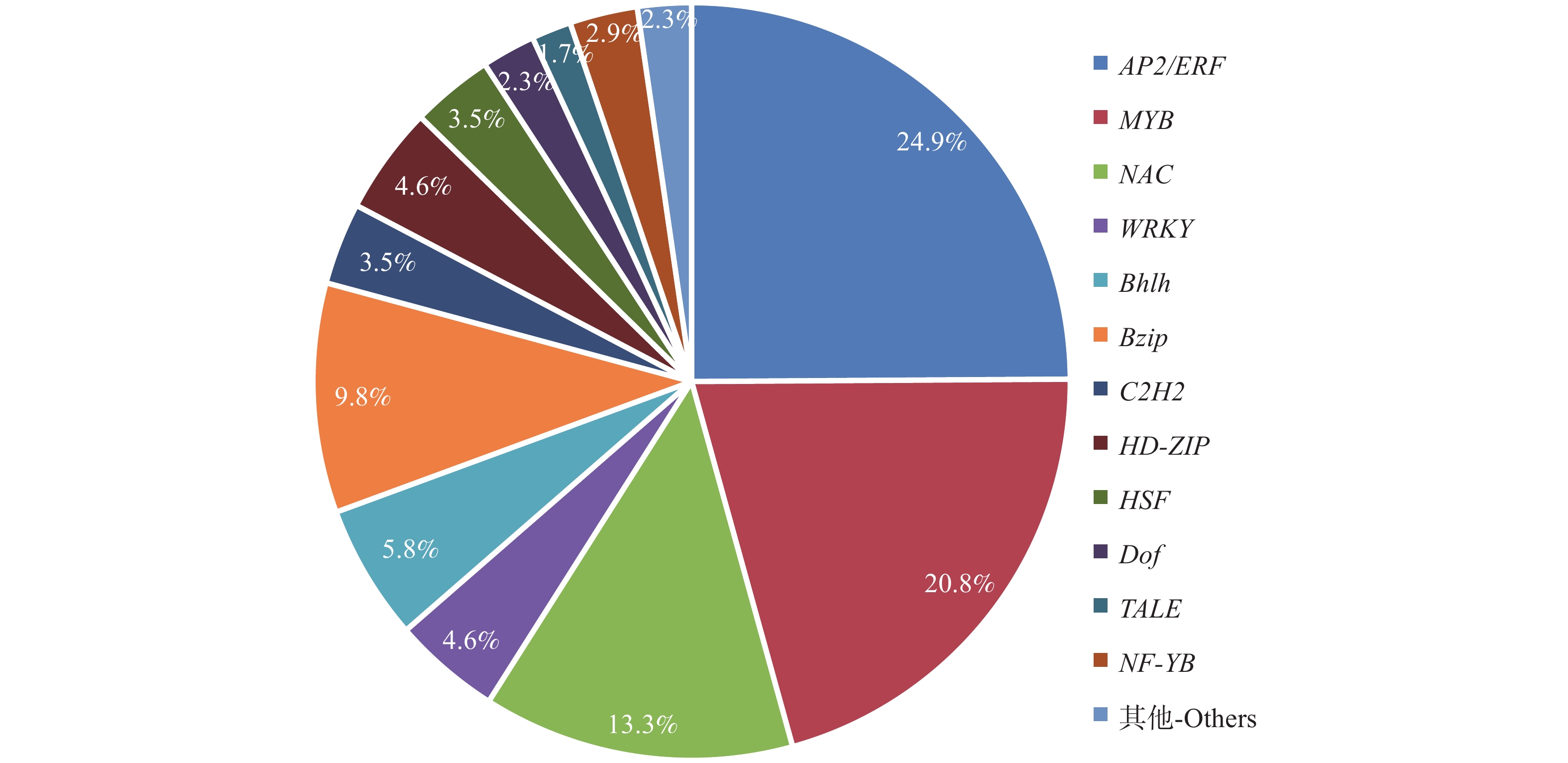
由于GO富集分析发现差异表达基因在DNA结合转录因子活性条目中高度富集,且也有许多文献中报道了多种转录因子在共生固氮中发挥重要作用,因此我们进一步对响应早期共生固氮的转录因子进行筛选,共筛选出172个差异表达的转录因子。将筛选出的差异表达的转录因子与植物转录因子数据库PlantTFDB(https://planttfdb.gao-lab.org/index.php)进行比对后,统计各转录因子家族的数量和类型,结果表明差异表达的转录因子主要包括AP2/ERF(36个)、MYB(23个,包含MYB-相关的)、NAC(8个)、WRKY(10个)、bHLH(17个)等家族(图3)。在差异表达的转录因子中,AP2/ERF家族成员的转录因子数量最多,且AP2/ERF家族转录因子已被报道参与植物生长发育、胁迫响应、激素信号转导等多个重要的生物学过程,因此我们推测AP2/ERF转录因子可能在大豆早期共生固氮中发挥重要作用。由于AP2/ERF家族可根据AP2/ERF结构域的数量以及是否含有其他结构域进一步分为AP2、ERF、DREB、RAV和Soloist5个亚家族,分别将差异表达的36个AP2/ERF家族成员与已鉴定的大豆各亚家族成员进行比对,发现差异表达的AP2/ERF家族成员中有34个来自ERF亚家族,1个(Glyma.01G087500)来自RAV亚家族,1个(Glyma.12G073300)来自AP2亚家族。
![]() 注:其它中包括与转录因子数据库比对不上的推测转录因子以及个数在2以下的转录因子。Note: Others include putative transcription factors that do not match the transcription factor database and transcription factor with a number of less than 2.图 3 差异表达的转录因子类型及数量统计Fig. 3 Differentially expressed transcription factor type and quantity statistics
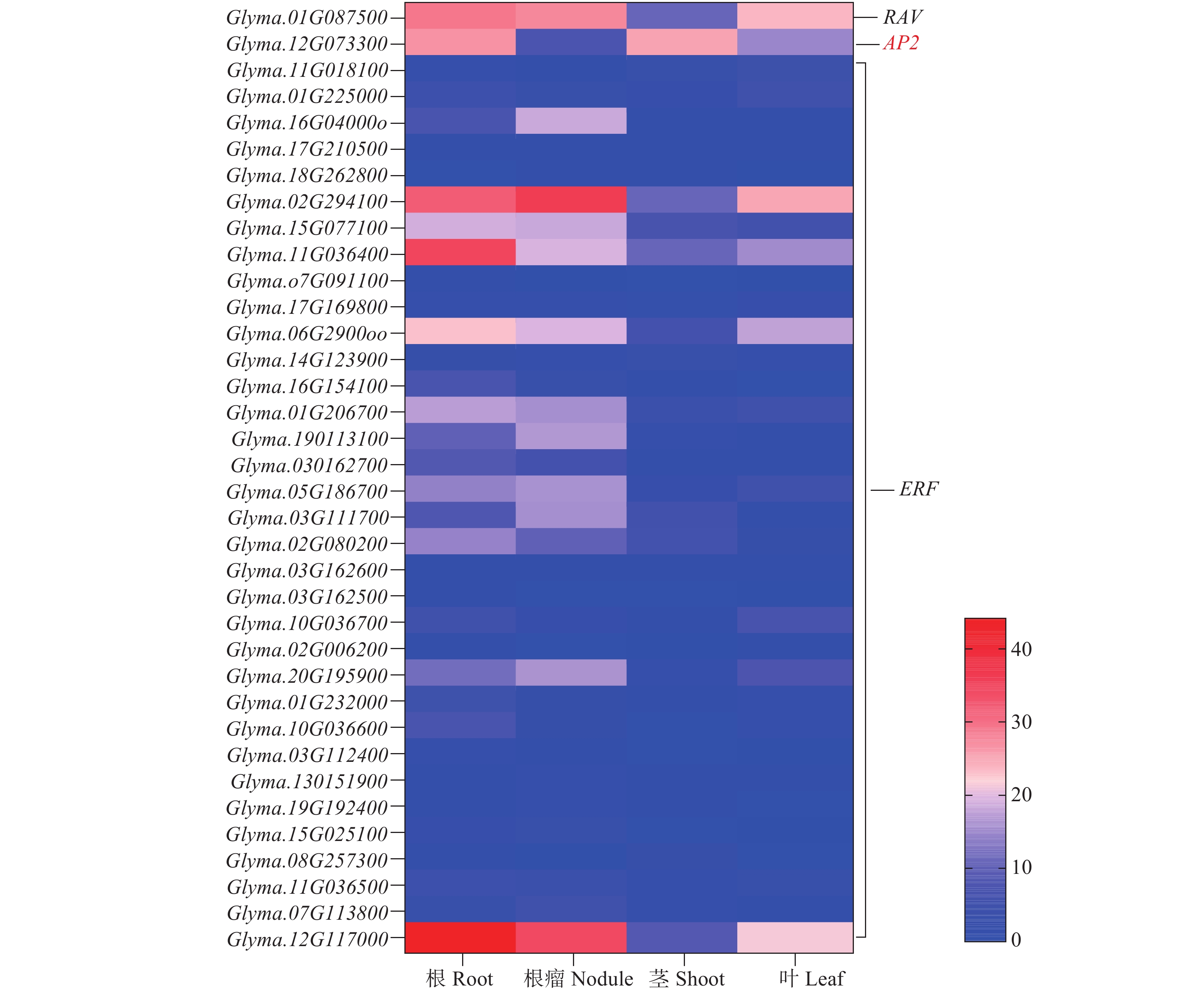
注:其它中包括与转录因子数据库比对不上的推测转录因子以及个数在2以下的转录因子。Note: Others include putative transcription factors that do not match the transcription factor database and transcription factor with a number of less than 2.图 3 差异表达的转录因子类型及数量统计Fig. 3 Differentially expressed transcription factor type and quantity statistics为了了解响应早期共生固氮的AP2/ERF转录因子各亚家族成员基因表达特点,利用已公开的植物RNA-seq在线数据库(https://plantrnadb.com/)[28]下载36个AP2/ERF转录因子家族成员在根(Root)、根瘤(nodule)、地上部(shoot)、叶(leaf)等不同组织中的基因表达相关数据,并使用GrapPad Pism 8制作基因表达热图,对AP2/ERF各亚家族成员在不同组织中的表达模式进行分析。结果表明,相较于其它成员,RAV亚家族的Glyma.01G087500在根、根瘤和叶片中的表达水平比较高,AP2亚家族的Glyma.12G073300在根和地上部的水平都比较高,ERF亚家族中的Glyma.02G294100和Glyma.12G117000在根、根瘤和叶片中的表达水平比较高,Glyma.11G036400和Glyma.06G290000在根中表达水平较高,而Glyma.17G210500、Glyma.18G262800、Glyma.07G091100、Glyma.17G169800、Glyma.03G162600、Glyma.03G162500、Glyma.02G006200、Glyma.03G112400、Glyma.13G151900、Glyma.19G192400在4个组织中几乎不表达,表明这10个基因可能为诱导表达基因(图4)。
![]() 图 4 AP2/ERF家族成员基因在不同组织中的表达热图Fig. 4 Expression heat map of AP2/ERF family members in different tissues
图 4 AP2/ERF家族成员基因在不同组织中的表达热图Fig. 4 Expression heat map of AP2/ERF family members in different tissues2.4 AP2/ERF家族进化分析
AP2/ERF转录因子家族中的ERN在协调诱导蒺藜苜蓿和百脉根根瘤菌侵染和根瘤器官发生方面发挥重要作用[18,31 − 32]。为了解蒺藜苜蓿中的ERN1/ERN2、百脉根中的ERN及参与早期共生固氮的AP2/ERF家族成员之间的进化关系,利用蛋白序列构建了系统进化树(图5),结果表明根据遗传距离,大豆中AP2/ERF家族成员可以聚为三类,第Ⅰ分支中的Glyma.19G113100、Glyma.16G040000、Glyma.16G154100、Glyma.18G262800、Glyma.07G091100等18个基因与MtERN1/2和LjERN亲缘关系较近,其中Glyma.16G040000、Glyma.16G154100、Glyma.18G262800与MtERN1/2和LjERN的序列相似性达到86%及以上,可能也在诱导根瘤菌侵染和根瘤器官发生中发挥作用,而第Ⅱ分支中的9个基因、第Ⅲ分支中的9个基因则与MtERN1/2和LjERN亲缘关系较远。
2.5 qRT-PCR验证
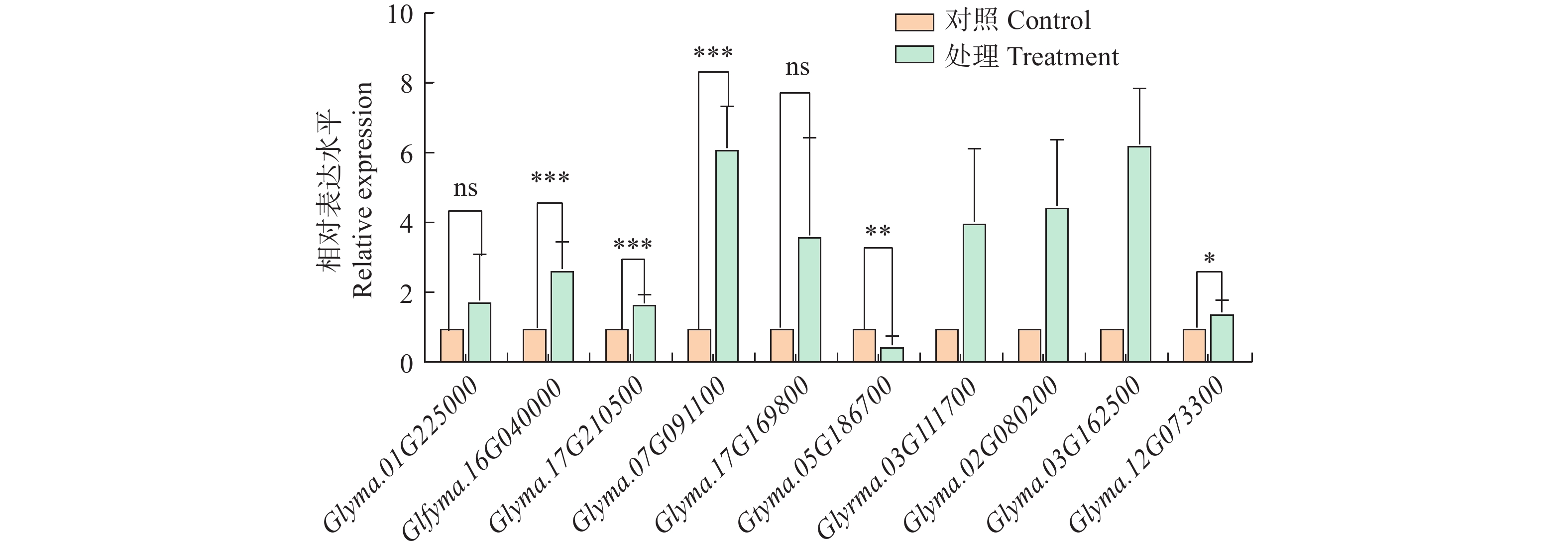
使用ELF-1b作为内参基因,在差异表达的AP2/ERF家族成员中挑选了10个基因通过qRT-PCR验证基因表达量变化趋势是否与转录组数据一致,根据转录组数据Glyma.01G225000、Glyma.16G040000、Glyma.17G210500、Glyma.07G091100、Glyma.17G169800、Glyma.12G073300这6个基因显示表达上调,Glyma.05G186700、Glyma.03G111700、Glyma.02G080200、Glyma.03G162500这4个基因显示表达下调。验证结果如图6所示,Glyma.01G225000、Glyma.16G040000、Glyma.17G210500、Glyma.07G091100、Glyma.17G169800、Glyma.05G186700、Glyma.12G073300这7个基因的表达情况与转录组数据趋势一致,其中Glyma.16G040000、Glyma.17G210500、Glyma.07G091100、Glyma.05G186700、Glyma.12G073300这5个基因的表达在不同程度上被显著调控。qRT-PCR验证结果与转录组测序结果相符,表明AP2/ERF家族基因确实受到根瘤菌侵染的调控。
![]() 注:*:P<0.05,**:P<0.01,***:P<0.001;ns表示差异不显著。Note: *: P<0.05, **: P<0.01, ***: P<0.001 ; ns indicates the difference is not significant.图 6 AP2/ERF家族成员基因在接种根瘤菌1 d 后的相对表达水平Fig. 6 Relative expression levels of AP2 / ERF family members 1 d after inoculation with rhizobia
注:*:P<0.05,**:P<0.01,***:P<0.001;ns表示差异不显著。Note: *: P<0.05, **: P<0.01, ***: P<0.001 ; ns indicates the difference is not significant.图 6 AP2/ERF家族成员基因在接种根瘤菌1 d 后的相对表达水平Fig. 6 Relative expression levels of AP2 / ERF family members 1 d after inoculation with rhizobia3 讨论与结论
本研究利用RNA-Seq技术对不接种根瘤菌与接种根瘤菌1 d后的大豆材料的根组织进行转录组测序分析,来挖掘大豆根中响应早期共生固氮的关键基因。研究发现,接种根瘤菌后1分钟内即可发生根瘤菌在根毛上的附着,并诱导钙离子内流和膜去极化;随后在12 h内这些初步的细胞信号事件促使根毛发生变形和卷曲,在24 h内根瘤菌侵入根毛细胞并形成侵染线;接着在24 ~ 96 h内表皮下的皮层细胞开始分裂,与此同时根瘤菌随着侵染线伸长往皮层延伸[33]。本研究共筛选出了328个受根瘤菌侵染表达显著上调的基因和496个表达显著下调的基因,10个基因中有7个(70%)的qPCR结果与转录谱数据趋势一致(图6),表明RNA-Seq数据可靠。
在差异表达基因中Glyma.01G028500、Glyma.09G187000、Glyma.02G311000、Glyma.01G241900、Glyma.06G184400这5个基因在共生固氮中的作用已有文献报道。值得注意的是,Glyma.09G187000(GmFWL1)响应根瘤菌侵染在根毛中表达,且随后在根瘤发育过程中被强烈诱导表达,表明其在共生固氮早期和结瘤过程中都发挥了作用。RNAi沉默GmFWL1可显著减少根瘤数量并减小细胞核大小,由于GmFWL1蛋白定位于质膜,且植物中FWL基因家族具有强保守性,表明GmFWL1可能是通过细胞级联反应来影响细胞核结构[29]。GmFWL1蛋白是一种质膜微结构域相关蛋白,能够与remorin、prohibitins和flotillins等多种膜微结构域相关蛋白互作。例如,GmFWL1能和GmFLOT2/4(与蒺藜苜蓿结瘤过程中的一个主要调控因子MtFLOT4同源的蛋白质)互作在根毛细胞尖端形成微结构域蛋白质复合物,这种蛋白质复合物可以催化根瘤菌对根毛细胞的初始内吞渗透[34]。Glyma.01G241900(GmNMNa)在共生固氮早期阶段调控根瘤菌入侵和存活,其编码的蛋白质定位于核仁和线粒体,沉默GmNMNa导致结瘤数量和侵染细胞中的类菌体数量减少,类菌体中PHB的积累也明显减少,这可能是由于GmNMNa沉默影响了碳代谢(较少的类菌体和PHB),破坏根瘤线粒体功能,从而导致结瘤数量减少[30]。与前期研究结果一致,本研究中GmFWL1和GmNMNa的差异性表达为它们在共生固氮早期阶段发挥作用提供了有力的证据。
GO富集和KEGG富集分析结果表明,DEGs主要富集在转录调控、激素相关以及次生代谢产物合成等通路上。差异表达基因显著富集在转录调控上,与在早期共生固氮中为响应根瘤菌侵染、细胞转录因子活性升高来激活结瘤信号通路以诱导基因表达以及接种根瘤菌1 d后皮层细胞开始分裂的观点一致[23,35]。研究表明,赤霉素、乙烯、脱落酸等植物激素信号传导对于豆科植物-根瘤菌共生至关重要[36]。GA20氧化酶(GA20ox)是一种催化赤霉素合成过程中的限速因子,蒺藜苜蓿中的MtGA2ox10通过对赤霉素分解代谢的精准调控在根瘤菌侵染和根瘤发育过程中发挥重要作用,与本研究中差异表达基因显著富集在赤霉素20氧化酶活性上也是一致的[37]。
已有研究表明:脱落酸能够调控植物中萜类化合物、苯丙烷类化合物以及其他类型次生代谢产物的生物合成途径[38]。本研究表明:根瘤菌侵染在共生固氮早期显著影响根系中代谢产物的积累,尤其是二萜类化合物、异黄酮、萜类化合物骨架、苯丙烷类化合物、类黄酮等次生代谢产物。通过RNAi沉默查尔酮合成酶(Chalcone synthase,CHS)破坏类黄酮合成途径可抑制蒺藜苜蓿中根瘤的形成,并阻止根瘤对生长素转运的调控,降低其他的类黄酮化合物生物合成基因表达,也在不同程度上抑制结瘤,而外源施加类黄酮前体能挽救根瘤形成和类黄酮积累[39 − 40]。此外,转录因子在调控萜类合成方面具有重要作用,目前在植物中已经鉴定出参与萜类合成调控的MYB、AP2/ERF、WRKY、bHLH、NAC和bZIP等转录因子家族[41]。综合以上信息可以看出,具有转录调控功能的转录因子在早期共生固氮中发挥了重要作用。
大豆AP2/ERF转录因子家族成员的组织表达模式分析结果显示,AP2/ERF家族成员在不同组织中有不同程度的表达,RAV亚家族的Glyma.01G087500在根、根瘤和叶片中的表达水平比较高,AP2亚家族的Glyma.12G073300在根和地上部的水平都比较高,ERF亚家族中的Glyma.02G294100和Glyma.12G117000在根、根瘤和叶片中的表达水平比较高,Glyma.11G036400和Glyma.06G290000在根中表达水平较高,而Glyma.17G210500、Glyma.18G262800、Glyma.07G091100、Glyma.17G169800、Glyma.03G162600、Glyma.03G162500、Glyma.02G006200、Glyma.03G112400、Glyma.13G151900、Glyma.19G192400在4个组织中几乎不表达,表明这10个基因可能为诱导表达基因,接种根瘤菌后其表达水平发生变化主要是由于受到根瘤菌侵染调控。
ERN1是百脉根根瘤菌侵染的关键调节因子,也是CCaMK/CYCLOPS复合体的直接靶标,其在表皮中的表达依赖于CYCLOPS[42]。进化分析结果表明大豆中AP2/ERF家族成员可根据与MtERN1/2和LjERN亲缘关系的远近聚为三个分支。第Ⅰ分支中的Glyma.19G113100、Glyma.16G040000、Glyma.16G154100、Glyma.18G262800、Glyma.07G091100等18个基因与MtERN1/2和LjERN亲缘关系较近,其中Glyma.16G040000、Glyma.16G154100、Glyma.18G262800与MtERN1/2和LjERN的序列相似性达到86%及以上,表明这4个基因可能也在诱导根瘤菌侵染和根瘤器官发生中发挥作用,而与MtERN1/2和LjERN亲缘关系较远的第Ⅱ分支、第Ⅲ分支中的转录因子在早期共生固氮中发挥何种作用仍有待进一步探究。
-
![]()
注:Rich factor表示位于GO条目中的差异基因个数与总基因数的比值,其大小与富集程度呈正比,颜色与P值大小有关。
Note: Rich factor represents the ratio of the number of differentially expressed genes in the GO entries to the total number of genes, which is proportional to the degree of enrichment, and the color is related to the size of the P value.
图 2 差异表达基因的GO富集散点图(A)和差异表达基因的KEGG富集散点图(B)
Figure 2. GO enrichment scatter plot of differentially expressed genes (A) and KEGG enrichment scatter plot of differentially expressed genes (B)
![]()
注:其它中包括与转录因子数据库比对不上的推测转录因子以及个数在2以下的转录因子。
Note: Others include putative transcription factors that do not match the transcription factor database and transcription factor with a number of less than 2.
图 3 差异表达的转录因子类型及数量统计
Figure 3. Differentially expressed transcription factor type and quantity statistics
![]()
图 4 AP2/ERF家族成员基因在不同组织中的表达热图
Figure 4. Expression heat map of AP2/ERF family members in different tissues
![]()
注:*:P<0.05,**:P<0.01,***:P<0.001;ns表示差异不显著。
Note: *: P<0.05, **: P<0.01, ***: P<0.001 ; ns indicates the difference is not significant.
图 6 AP2/ERF家族成员基因在接种根瘤菌1 d 后的相对表达水平
Figure 6. Relative expression levels of AP2 / ERF family members 1 d after inoculation with rhizobia
表 1 qRT-PCR引物名称及序列
Table 1 Primer name and sequence of qRT-PCR
引物名称
Primer name序列(5'-3')
Sequence(5'-3')引物名称
Primer name序列(5'-3')
Sequence(5'-3')qRT-Gm01G225000-F GGAGGGAATGCGAGACAGAA qRT-Gm03G111700-F TTGAGATACCCGTAACGACGC qRT-Gm01G225000-R TGTTGTTGTGGGGCAATTCG qRT-Gm03G111700-R CTCCGCCCAATTCTCTGTCA qRT-Gm16G040000-F CCAAACCTGCAGCAACAGAAAT qRT-Gm02G080200-F TAGCATCTCAAAGGGTGTCCTG qRT-Gm16G040000-R ATCTCAGCAACCCATCTCCC qRT-Gm02G080200-R TCCGTGAACCGCATAAACTCA qRT-Gm17G210500-F GGGAGTAAGACAGCGCCAAT qRT-Gm03G162500-F TCATGGGAAGAGCTGTTTTCG qRT-Gm17G210500-R GCAGCCTCTGCTGTCTCAAA qRT-Gm03G162500-R ATTTTTCTTCAACGTGGCCGT qRT-Gm07G091100-F AGGCAAACTCAGAAACAAGGAA qRT-Gm12G073300-F GACAGCGAAGTAGAAGCTGCAAG qRT-Gm07G091100-R GTGGCATCAGGGTCAGTGTA qRT-Gm12G073300-R CTGCCCATTGGGAAAGAAATTAGG qRT-Gm17G169800-F AGGGTGAAACTGAGACGACG qRT-ELF1b-F GTTGAAAAGCCAGGGGACA qRT-Gm17G169800-R GAAAACGGCGGTGTCGTAAG qRT-ELF1b-R TCTTACCCCTTGAGCGTGG qRT-Gm05G186700-F AACGACGTTGACCAGAGTTACA qRT-Gm05G186700-R AAAGCAGAGATCATGGCTGACA  下载: 导出CSV
下载: 导出CSV
表 2 qRT-PCR反应体系
Table 2 qRT-PCR reaction system
试剂
Reagent用量
Consumption/μLcDNA 1 Primer F 0.2 Primer R 0.2 ChamQ Blue Universal SYBR qPCR Master Mix 5 ddH2O 3.6
下载: 导出CSV
表 3 qRT-PCR反应程序
Table 3 qRT-PCR reaction procedure
温度
Temperature/ ℃时间
Time/s95 30 95 10 $\Biggr\} $ 40 cycles 60 30
下载: 导出CSV
表 4 测序数据质量分析
Table 4 Quality analysis of sequencing data
样品编号
Sample code原始数据
Raw reads有效数据
Valid reads有效数据占比
Valid ratio/%质量值≥30的碱基占比
Q≥30/%GC含量占比
GC content/%NN_1d_CK1 41981696 40909042 97.44 96.98 44.5 NN_1d_CK2 42443112 41316398 97.35 97.28 44.5 NN_1d_CK3 43652536 42547784 97.47 97.32 44.5 NN_1d_Bj1 41971824 40604942 96.74 97.31 44.5 NN_1d_Bj2 41274128 40152166 97.28 97.25 44.5 NN_1d_Bj3 44167656 43034476 97.43 97.33 44.5
下载: 导出CSV
-
[1] YANG J, LAN L Y, JIN Y, et al. Mechanisms underlying legume-rhizobium symbioses[J]. Journal of Integrative Plant Biology,2022,64(2):244−267. DOI: 10.1111/jipb.13207
[2] DÉNARIÉ J, CULLIMORE J. Lipo-oligosaccharide nodulation factors: a minireview new class of signaling molecules mediating recognition and morphogenesis[J]. Cell,1993,74(6):951−954. DOI: 10.1016/0092-8674(93)90717-5
[3] PETERS N K, FROST J W, LONG S R. A plant flavone, luteolin, induces expression of Rhizobium meliloti nodulation genes[J]. Science,1986,233(4767):977−980. DOI: 10.1126/science.3738520
[4] REDMOND J W, BATLEY M, DJORDJEVIC M A, et al. Flavones induce expression of nodulation genes in Rhizobium[J]. Nature,1986,323:632−635. DOI: 10.1038/323632a0
[5] SHUMILINA J, SOBOLEVA A, ABAKUMOV E, et al. Signaling in legume-rhizobia symbiosis[J]. International Journal of Molecular Sciences,2023,24(24):17397. DOI: 10.3390/ijms242417397
[6] OLDROYD G E D. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants[J]. Nature Reviews Microbiology,2013,11(4):252−263. DOI: 10.1038/nrmicro2990
[7] GUAN D, STACEY N, LIU C W, et al. Rhizobial infection is associated with the development of peripheral vasculature in nodules of Medicago truncatula[J]. Plant Physiology,2013,162(1):107−115. DOI: 10.1104/pp.113.215111
[8] OLDROYD G E D, MURRAY J D, POOLE P S, et al. The rules of engagement in the legume-rhizobial symbiosis[J]. Annual Review of Genetics,2011,45:119−144. DOI: 10.1146/annurev-genet-110410-132549
[9] MURRAY J D. Invasion by invitation: rhizobial infection in legumes[J]. Molecular Plant-Microbe Interactions,2011,24(6):631−639. DOI: 10.1094/MPMI-08-10-0181
[10] 张雪, 邱丽娟, 阎哲. 豆科植物结瘤自调控分子机制研究进展[J]. 土壤与作物,2022,11(1):1−9. DOI: 10.11689/j.issn.2095-2961.2022.01.001 ZHANG X, QIU L J, YAN Z. Research progress on molecular mechanisms of autoregulation of nodulation in legumes[J]. Soils and Crops,2022,11(1):1−9. DOI: 10.11689/j.issn.2095-2961.2022.01.001
[11] ANTOLÍN-LLOVERA M, RIED M K, PARNISKE M. Cleavage of the symbiosis receptor-like kinase ectodomain promotes complex formation with nod factor receptor 5[J]. Current Biology,2014,24(4):422−427. DOI: 10.1016/j.cub.2013.12.053
[12] GLEASON C, CHAUDHURI S, YANG T B, et al. Nodulation independent of rhizobia induced by a calcium-activated kinase lacking autoinhibition[J]. Nature,2006,441(7097):1149−1152. DOI: 10.1038/nature04812
[13] LÉVY J, BRES C, GEURTS R, et al. A putative Ca2+ and calmodulin-dependent protein kinase required for bacterial and fungal symbioses[J]. Science,2004,303(5662):1361−1364. DOI: 10.1126/science.1093038
[14] YANO K, YOSHIDA S, MÜLLER J, et al. CYCLOPS, a mediator of symbiotic intracellular accommodation[J]. Proceedings of the National Academy of Sciences of the United States of America,2008,105(51):20540−20545.
[15] SINGH S, KATZER K, LAMBERT J, et al. CYCLOPS, a DNA-binding transcriptional activator, orchestrates symbiotic root nodule development[J]. Cell Host and Microbe,2014,15(2):139−152.
[16] FONOUNI-FARDE C, TAN S, BAUDIN M, et al. DELLA-mediated gibberellin signalling regulates Nod factor signalling and rhizobial infection[J]. Nature Communications,2016,7:12636. DOI: 10.1038/ncomms12636
[17] JIN Y, LIU H, LUO D X, et al. DELLA proteins are common components of symbiotic rhizobial and mycorrhizal signalling pathways[J]. Nature Communications,2016,7:12433. DOI: 10.1038/ncomms12433
[18] CERRI M R, WANG Q H, STOLZ P, et al. The ERN1 transcription factor gene is a target of the CCaMK/CYCLOPS complex and controls rhizobial infection in Lotus japonicus[J]. The New Phytologist,2017,215(1):323−337. DOI: 10.1111/nph.14547
[19] JOURNET E P, EL-GACHTOULI N, VERNOUD V, et al. Medicago truncatula ENOD11: a novel RPRP-encoding early nodulin gene expressed during mycorrhization in arbuscule-containing cells[J]. Molecular Plant-Microbe Interactions,2001,14(6):737−748. DOI: 10.1094/MPMI.2001.14.6.737
[20] HIRSCH S, KIM J, MUÑOZ A, et al. GRAS proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula[J]. The Plant Cell,2009,21(2):545−557. DOI: 10.1105/tpc.108.064501
[21] ANDRIANKAJA A, BOISSON-DERNIER A, FRANCES L, et al. AP2-ERF transcription factors mediate nod factor–dependent Mt ENOD11 activation in root hairs via a novel cis-regulatory motif[J]. The Plant Cell,2007,19(9):2866−2885. DOI: 10.1105/tpc.107.052944
[22] MARSH J F, RAKOCEVIC A, MITRA R M, et al. Medicago truncatula NIN is essential for rhizobial-independent nodule organogenesis induced by autoactive calcium/calmodulin-dependent protein kinase[J]. Plant Physiology,2007,144(1):324−335. DOI: 10.1104/pp.106.093021
[23] LIU C W, BREAKSPEAR A, GUAN D, et al. NIN acts as a network hub controlling a growth module required for rhizobial infection[J]. Plant Physiology,2019,179(4):1704−1722. DOI: 10.1104/pp.18.01572
[24] MADSEN L H, TIRICHINE L, JURKIEWICZ A, et al. The molecular network governing nodule organogenesis and infection in the model legume Lotus japonicus[J]. Nature Communications,2010,1(1):10. DOI: 10.1038/ncomms1009
[25] HAYASHI S, REID D E, LORENC M T, et al. Transient Nod factor-dependent gene expression in the nodulation-competent zone of soybean (Glycine max[L. ]Merr. ) roots[J]. Plant Biotechnology Journal,2012,10(8):995−1010. DOI: 10.1111/j.1467-7652.2012.00729.x
[26] YUAN S L, LI R, CHEN S L, et al. RNA-Seq analysis of differential gene expression responding to different Rhizobium strains in soybean (Glycine max) roots[J]. Frontiers in Plant Science,2016,7:721.
[27] 祁平, 郑凯杰, 赵晓宇, 等. 根瘤菌侵染早期大豆根系的转录组分析[J]. 大豆科学,2021,40(3):289−298. DOI: 10.11861/j.issn.1000-9841.2021.03.0289 QI P, ZHENG K J, ZHAO X Y, et al. Transcriptome analysis of soybean root system in the early stage of Rhizobium infection[J]. Soybean Science,2021,40(3):289−298. DOI: 10.11861/j.issn.1000-9841.2021.03.0289
[28] YU Y M, ZHANG H, LONG Y P, et al. Plant Public RNA-seq Database: a comprehensive online database for expression analysis of ~45 000 plant public RNA-Seq libraries[J]. Plant Biotechnology Journal,2022,20(5):806−808. DOI: 10.1111/pbi.13798
[29] LIBAULT M, ZHANG X C, GOVINDARAJULU M, et al. A member of the highly conserved FWL (tomato FW 2.2-like) gene family is essential for soybean nodule organogenesis[J]. The Plant Journal,2010,62(5):852−864. DOI: 10.1111/j.1365-313X.2010.04201.x
[30] LIBAULT M, GOVINDARAJULU M, BERG R H, et al. A dual-targeted soybean protein is involved in Bradyrhizobium japonicum infection of soybean root hair and cortical cells[J]. Molecular Plant-Microbe Interactions,2011,24(9):1051−1060. DOI: 10.1094/MPMI-12-10-0281
[31] MIDDLETON P H, JAKAB J, PENMETSA R V, et al. An ERF transcription factor in Medicago truncatula that is essential for Nod factor signal transduction[J]. The Plant Cell,2007,19(4):1221−1234. DOI: 10.1105/tpc.106.048264
[32] CERRI M R, FRANCES L, KELNER A, et al. The symbiosis-related ERN transcription factors act in concert to coordinate rhizobial host root infection[J]. Plant Physiology,2016,171(2):1037−1054.
[33] BRECHENMACHER L, KIM M Y, BENITEZ M, et al. Transcription profiling of soybean nodulation by Bradyrhizobium japonicum[J]. Molecular Plant-Microbe Interactions,2008,21(5):631−645. DOI: 10.1094/MPMI-21-5-0631
[34] QIAO Z Z, BRECHENMACHER L, SMITH B, et al. The GmFWL1 (FW2-2-like) nodulation gene encodes a plasma membrane microdomain-associated protein[J]. Plant, Cell and Environment,2017,40(8):1442−1455.
[35] BREAKSPEAR A, LIU C W, ROY S, et al. The root hair "infectome" of Medicago truncatula uncovers changes in cell cycle genes and reveals a requirement for auxin signaling in rhizobial infection[J]. The Plant Cell,2015,26(12):4680−4701.
[36] FERGUSON B J, MATHESIUS U. Phytohormone regulation of legume-rhizobia interactions[J]. Journal of Chemical Ecology,2014,40(7):770−790. DOI: 10.1007/s10886-014-0472-7
[37] CHU X T, SU H N, HAYASHI S, et al. Spatiotemporal changes in gibberellin content are required for soybean nodulation[J]. The New Phytologist,2022,234(2):479−493. DOI: 10.1111/nph.17902
[38] 甄梦缘, 王丽芝, 孙超. 脱落酸及其调控植物次生代谢产物生物合成的研究进展[J]. 天津中医药大学学报,2024,43(3):259−267. ZHEN M Y, WANG L Z, SUN C. Research progress of abscisic acid and its regulation on biosynthesis of plant secondary metabolites[J]. Journal of Tianjin University of Traditional Chinese Medicine,2024,43(3):259−267.
[39] WASSON A P, PELLERONE F I, MATHESIUS U. Silencing the flavonoid pathway in Medicago truncatula inhibits root nodule formation and prevents auxin transport regulation by rhizobia[J]. The Plant Cell,2006,18(7):1617−1629. DOI: 10.1105/tpc.105.038232
[40] ZHANG J, SUBRAMANIAN S, STACEY G, et al. Flavones and flavonols play distinct critical roles during nodulation of Medicago truncatula by Sinorhizobium meliloti[J]. The Plant Journal,2009,57(1):171−183. DOI: 10.1111/j.1365-313X.2008.03676.x
[41] 董燕梅, 张文颖, 凌正一, 等. 转录因子调控植物萜类化合物生物合成研究进展[J]. 植物学报,2020,55(3):340−350. DOI: 10.11983/CBB19186 DONG Y M, ZHANG W Y, LING Z Y, et al. Advances in transcription factors regulating plant terpenoids biosynthesis[J]. Chinese Bulletin of Botany,2020,55(3):340−350. DOI: 10.11983/CBB19186
[42] CHAKRABORTY S, VALDÉS-LÓPEZ O, STONOHA-ARTHER C, et al. Transcription factors controlling the Rhizobium-legume symbiosis: integrating infection, organogenesis and the abiotic environment[J]. Plant and Cell Physiology,2022,63(10):1326−1343. DOI: 10.1093/pcp/pcac063
-
其他相关附件
-
DOCX格式
黄慧超 附表1 点击下载(20KB)
-

计量
- 文章访问数: 16
- HTML全文浏览量: 0
- PDF下载量: 2
